Living with primary immunodeficiency (PI) can be challenging, but you’re not alone—many people with PI lead full and active lives. With the right support and resources, you can, too.
Be a hero for those with PI. Change lives by promoting primary immunodeficiency (PI) awareness and taking action in your community through advocacy, donating, volunteering, or fundraising.
Whether you’re a clinician, researcher, or an individual with primary immunodeficiency (PI), IDF has resources to help you advance the field. Get details on surveys, grants, and clinical trials.
ALPS is a rare genetic disorder in which lymphocytes, a type of white blood cell, increase and accumulate in the spleen and lymph nodes. This is due to the failure of the mechanism that normally causes lymphocytes to die naturally.
Overview
ALPS was also known as the Canale-Smith syndrome before its genetic causes were discovered and inheritance patterns were understood.
ALPS is due to genetic variants in a gene coding for a specific protein called FAS. It is associated with childhood-onset increases in the size of lymph nodes (lymphadenopathy) and spleen (splenomegaly), as well as decreases in red blood cells, platelets, and, occasionally, neutrophils, as well as an increased lifetime risk of lymphoma. More than 1,000 individuals with ALPS-FAS and their relatives have been identified and followed worldwide.
The normal response to infection by a pathogen is the multiplication of lymphocytes in the lymph nodes. When the rapid growth of these cells in the lymph nodes and spleen is uncontrolled, excessive, and harmful, it is described as a lymphoproliferative syndrome.
Lymphocytes are normally subject to programmed cell death (known as apoptosis), controlled by the FAS gene. If there is a change in the genetic code in the FAS gene, also known as a genetic variant, the lymphocyte control pathway no longer functions normally and there is an uncontrolled expansion of certain lymphocytes. This is the cause of most ALPS cases and, hence, the condition is known currently as ALPS-FAS. In very rare cases, the defect in lymphocytes is caused by variants in other genes involved in the lymphocyte control pathway.
Chapter 17: Autoimmune Lymphoproliferative Syndrome and Related Disorders
ALPS can be diagnosed early in childhood when the child presents with chronically enlarged lymph nodes in the neck, groin, and armpits. ALPS is also often associated with an enlarged spleen. Other early presentations include fatigue, pallor, or jaundice (all the result of anemia), unusual bruising, frequent nosebleeds (due to low platelets), and frequent infections (due to low neutrophils).
“Always sick?” flyer
Use this printable flyer to raise awareness of PI in your community or share it with your healthcare provider if you think you may have PI.
ALPS can be difficult to diagnose because its symptoms and laboratory results overlap with a number of other common childhood diseases including childhood cancers, such as lymphoma and leukemia, and chronic viral infections, such as infectious mononucleosis, commonly known as mono.
ALPS is diagnosed when manifestations of autoimmunity are present along with two other factors: low blood cell counts including low hemoglobin, low platelets, and/or low neutrophils, and enlarged lymph nodes and/or spleen. The lymph nodes and spleen become enlarged because they are key lymphocyte storage and production sites. An enlarged liver (hepatomegaly) is also seen in 30-40% of individuals with ALPS.
Because ALPS is a genetic disorder, a family history of ALPS or other lymphoproliferative disease is also considered when making the diagnosis.
Failure of FAS-mediated cell death (apoptosis) can sometimes be detected with specialized laboratory tests, but very few laboratories are equipped to run these tests and the results are subject to some degree of uncertainty. Instead, certain biomarkers in the blood are strongly associated with ALPS, such as increased immunoglobulins, elevated serum vitamin B12 levels, and particular proteins including interleukin 10 (IL-10) and soluble FAS ligand (sFASL).
In many cases, antibodies that attack the individual’s own body (known as autoantibodies), such as antibodies to normal red blood cells, may also be present. Various forms of autoimmune cytopenias (low blood cell count) lead to anemia (low red blood cells that carry oxygen), thrombocytopenia (low platelets that normally help blood to clot at sites of injury), and/or neutropenia (low neutrophils that fight bacteria) and can be a serious and continuous problem for individuals with ALPS, especially young children.
Other autoimmune complications that are less common but possible with ALPS include glomerulonephritis (inflammation of the kidneys); autoimmune hepatitis (inflammation of the liver caused by immune attack); vasculitis (inflammation and destruction of blood vessels); uveitis (inflammation of the colored part of the eye); and, rarely, chronic pancreatitis (inflammation of the pancreas).
Read the latest research
Read the latest research on ALPS on PubMed. Note that not all publications listed in PubMed are freely available; some require a subscription to the publishing journal.
Two of the key manifestations of the disease, lymphoproliferation and splenomegaly, are especially difficult to treat but may not always cause severe harm without treatment. Treatments for ALPS usually focus on the complications due to autoimmune conditions associated with ALPS, especially severe autoimmune cytopenias, which can be life threatening.
Splenectomy (removal of spleen by surgery) was often used to treat the autoimmune cytopenias until the early 2000s. However, since then, spleen-sparing immunomodulatory regimens have become the mainstay of long-term therapy for these complications in individuals with ALPS. The shift away from splenectomy was driven by the observation that individuals with ALPS had a greater than normal risk of suffering a severe and potentially life-threatening infection after splenectomy.
The drugs used to treat autoimmune cytopenias include corticosteroids, mycophenolate mofetil (MMF), and rapamycin (also known as sirolimus). Rapamycin has been shown to reduce spleen and lymph node size over the long term. MMF does not reduce lymph node or spleen size and can cause hypogammaglobulinemia, which is a condition of a low level of immunoglobulins and may require immunoglobulin (Ig) replacement therapy.
Rapamycin has been used to resolve 90% of autoimmune symptoms as well as enlargement of lymph nodes and spleen. This drug may cause immune suppression leading to an increased risk for infections, though it has not been reported to cause hypogammaglobulinemia. Individuals taking rapamycin require careful monitoring as the drug can increase the risk of various cancers, cause mucositis (painful inflammation and ulceration of the digestive tract), diarrhea, hyperlipidemia (increased lipid/fat levels in blood), and slow the healing of wounds.
Corticosteroids can and should be used only as a short-term pulse therapy to address cytopenias and organ enlargement, and overall they have been met with limited success. Hematopoietic stem cell transplantation (HSCT) has also been used for some individuals. However, generally, HSCT is not advised because the good prognosis of most individuals with ALPS usually doesn’t warrant the challenges and risks associated with HSCT.
The spleen plays a critical role in fighting a common bacteria called pneumococci. If ALPS-related splenomegaly becomes severe enough, long-term rapamycin and short-term corticosteroids should be used to shrink the spleen. As noted above, splenectomy can result in a significant risk of sepsis with encapsulated bacteria, which is a lifethreatening condition in which tissues and organs are infected by bacteria and damaged by the body’s response to it. Therefore, individuals whose spleens have been removed should employ long-term antibiotic treatment and consult a healthcare provider immediately upon developing a fever. More individuals with ALPS-FAS have died due to infection developed many years after splenectomy than from any other cause, including lymphoma.
Management
Unlike many forms of primary immunodeficiency, ALPS itself does not significantly raise the risk of severe or frequent infection. Individuals who have undergone splenectomy should wear a medical alert bracelet, take prophylactic antibiotics, and seek immediate medical care for any fevers.
Complications associated with ALPS, including autoimmune hepatitis, vasculitis, uveitis, and others, should be managed just as they would be in people who do not have ALPS.
Because of its genetic nature, ALPS often affects more than one family member. Prompt recognition and treatment of any symptoms is important for optimal outcomes. Symptoms associated with ALPS are sometimes most severe in early childhood, when the lymphatic system is expanding, but may improve during adolescence and adulthood.
Individuals with ALPS have a higher risk of developing B cell lymphomas (Hodgkin and non-Hodgkin lymphoma), and they require frequent screening and monitoring for these cancers on a lifelong basis. Whenever individuals with ALPS develop systemic symptoms (like fever, night sweats, weight loss, or loss of appetite) and/or sudden focal enlargement of a group of their lymph nodes beyond their norm of clinical and seasonal variability, they should undergo further medical evaluations. These may include radiological imaging with computerized tomography (CT) and/or positron emission tomography (PET) scans followed by a biopsy of an appropriate lymph node(s). Routine frequent whole-body imaging on a yearly basis only for monitoring lymph nodes for lymphoma yields little benefit, and it is not recommended for individuals with ALPS.
In children, the enlargement of lymph nodes can be severe enough to be visible, resulting in potential anxiety and embarrassment. Shrinkage of the nodes for cosmetic reasons is not recommended, although attention to the child or young adult’s overall well-being and emotional concerns is equally vital. The nodes should be carefully watched for any sudden and significant changes in size, which should be promptly reported to the healthcare provider. The size of the lymph nodes and the spleen should also be regularly monitored through measurement by the medical team.
Although there are reports of successful use of rapamycin and MMF, the long-term toxicities of these therapies remain unknown. Through proper care, ALPS is usually manageable, and most individuals can have a relatively normal lifestyle and life span. However, this does not preclude the need for careful lifelong monitoring for possible progression to autoimmune and malignant diseases, particularly in children and young adults.
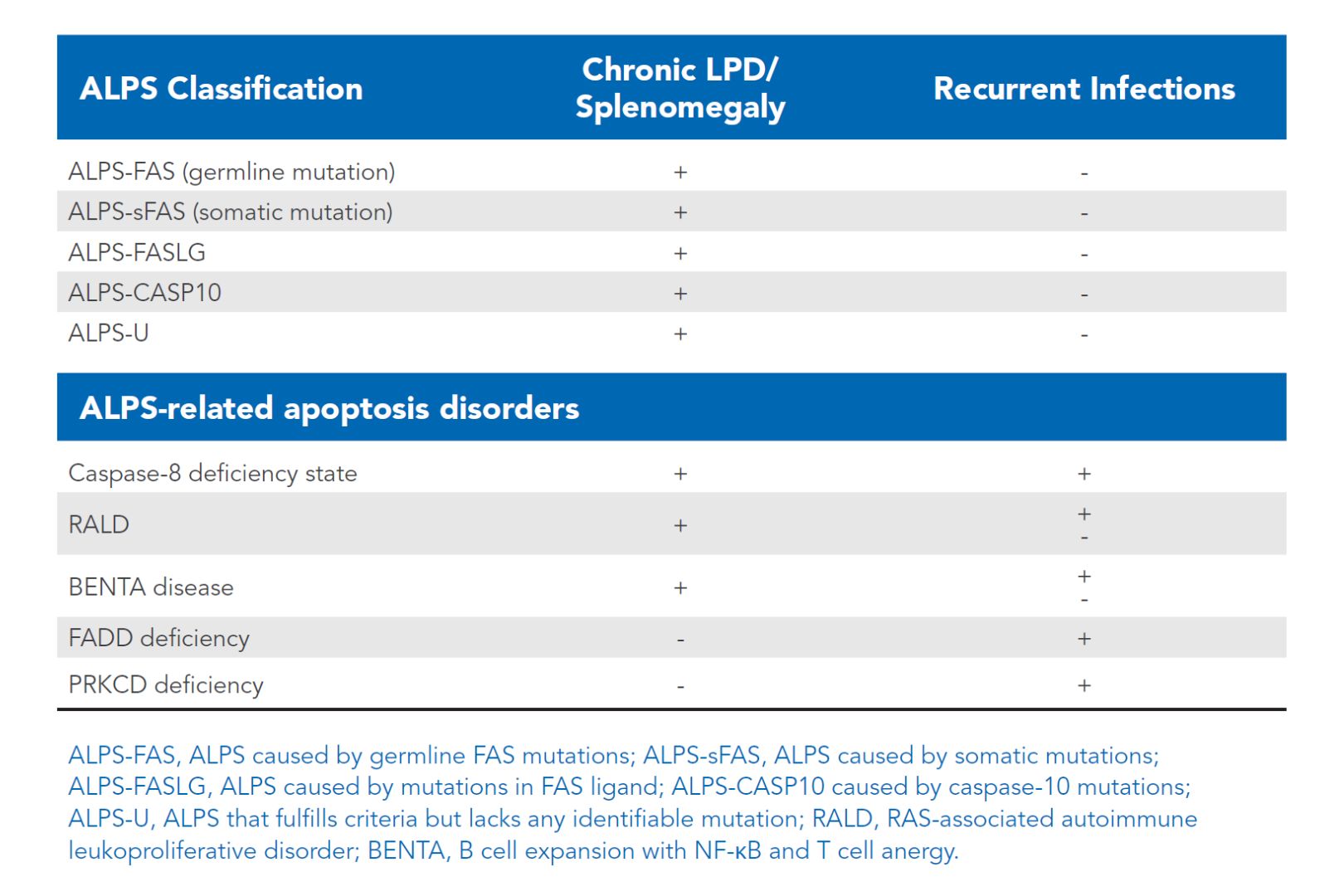
More recently, additional genetic variants have been discovered that mimic ALPS with similar clinical findings. For the sake of this material, they will be referred as ALPS related disorders (Table 17:1). Knowing the genetic basis of many of these lymphoproliferative disorders with autoimmune problems may facilitate better and more targeted treatment of these individuals.
Table 17:1 ALPS-related disorders (click table to enlarge).
References
Rao VK, Oliveira JB. How I treat autoimmune lymphoproliferative syndrome. Blood. 2011;118:5741–5751.
Price S, Shaw PA, Seitz A, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123;1989–1999.
Bonilla FA, Khan DA, Ballas ZK, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186-1205.e78.
Rao VK. Approaches to Managing Autoimmune Cytopenias in Novel Immunological Disorders with Genetic Underpinnings Like Autoimmune Lymphoproliferative Syndrome. Front Pediatr. 2015 Jul 21;3:65. doi: 10.3389/fped.2015.00065. eCollection 2015. Invited Review. PMID: 26258116.
Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, Chatila T, Crow YJ, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Puck J, Tang MLK, Tangye SG, Torgerson TR, Casanova JL, Sullivan KE. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2018 Jan;38(1):129-143. doi: 10.1007/s10875-017-0465-8. Epub 2017 Dec 11. PMID: 29226301
Diagnostic Center for Heritable Immunodeficiencies: https://www.cincinnatichildrens.org/service/d/dchi
This page contains general medical and/or legal information that cannot be applied safely to any individual case. Medical and/or legal knowledge and practice can change rapidly. Therefore, this page should not be used as a substitute for professional medical and/or legal advice. Additionally, links to other resources and websites are shared for informational purposes only and should not be considered an endorsement by the Immune Deficiency Foundation.